
The life sciences industry, which includes the production of pharmaceuticals and medical devices, faces unique challenges that have made it a late adopter of continuous improvement techniques such as Lean Six Sigma (LSS).
Most critical are severe government regulation, product approval phases that can span multiple years, and the imperative responsibility to ensure patient safety by delivering the highest level of product quality possible. This last component – safety, no matter how costly or inefficient — in particular has driven a high level of operational waste within many business units of life sciences companies.

Pharmaceutical companies in particular are feeling increased pressure to eliminate costs to compensate for market pressures coming from a recent patent cliff (an atypically high number of pharmaceutical patents reaching expiration within a short timeframe), decreased return on investment on research and development expenditures, and increased pressure from generic substitutes.
Those pressures have driven life sciences companies to turn to LSS methodologies within the past three to five years to identify and eliminate wasted costs from the value stream. While these methodologies have gained a stronger foothold in life sciences, their ability to uncover hidden wastes has been limited. The crucial question of “How much am I spending to deliver a quality product?” has largely remained unanswered.
Ineffective Cost Reduction Approaches
Lean has proven successful in some operational infrastructure aspects. 5S (sort, straighten, shine, standardize and sustain) has been widely implemented in labs, poka-yoke and other error-proofing methods have been included in inspection, and “pull” management of inventory levels is starting to be used for batching.
Many of the principles of Lean, however, are most effective in a high-volume, discrete-process manufacturing setting. Principles like one-piece flow and rapid iterative improvement have proven challenging to implement in such highly restricted environments as pharmaceuticals or medical devices. For example, since regulatory filings for life sciences companies are required for the vast majority of manufacturing process changes, the benefits of making small continuous improvements, as Lean encourages, are often outweighed by the costs of validating and documenting these proposed improvements.
The Food and Drug Administration (FDA) rolled out the process analytical technology (PAT) initiative in the last decade; the focus of PAT is to encourage FDA-regulated companies to manage product release and product stability testing through a set of critical process parameters that monitor a set of product critical quality attributes. While this certainly falls in line with core Six Sigma principles, few companies have invested the resources required to get such a program up and running. In fact, studies have shown that approximately only 10 percent of life sciences companies have implemented statistical process control (SPC), far below the general industry average. The more successful Six Sigma techniques have been project management based, with which companies have made strides in using the DMAIC process for root cause analysis with sustainable results.
A New Way of Looking at Costs
A popular LSS tool is the cost of poor quality (COPQ). A succinct definition of COPQ is any cost that would not be incurred if quality were perfect.
Some studies estimate the COPQ within life sciences at 25 percent to 35 percent of sales revenue, while industries such as semiconductors are only at 4 percent to 8 percent. Others have estimated that these costs can be 3 to 6 times larger than profit levels. This is a huge opportunity that has been largely untouched thus far.
The nature of the business of life sciences dictates that outgoing quality levels must be extremely high – unfortunately, almost without regard to cost or the resulting retail price to consumers. Therefore it comes as little surprise that the pharmaceutical and medical device industries score high marks for finished goods yields and customer defects, but often fail in comparison to other industries when operational efficiency metrics are compared.
The traditional view that life sciences has taken with regards to COPQ is to focus on the non-conformance aspect, which addresses both internal and external failure costs. This has resulted in quality being tested in to products, rather than built in upfront. When key parameters in manufacturing are not being monitored real-time in control charts, deficiencies in process are often hidden until final inspection, greatly raising the exposure to process waste and the potential for scrapped product.
In order to properly contain the scope of assessing COPQ, it needs to be treated as a strategic initiative like LSS, and staff must be trained accordingly. One reason often stated for the lack of executive management buy in for launching such an initiative is the inherent negative connotations that go with assessing and eliminating “poor” quality.
How can poor quality exist if finished goods yields are so high? Should blame be placed on the quality department for incurring these costs? Does improving these costs mean that the efforts of quality employees are no longer needed?
Cost of Poor Process and Product Quality (C3PQ)
A much more effective way of positioning a COPQ initiative is to explicitly state that the focus will be on eliminating process and product waste through the entire value stream. That is to say, increase the overall efficiency of quality-related processes by determining what is really value-added in the eyes of the customer as well as remain up-to-date on what non-value-added activities are absolutely mandated by regulatory agencies.
In this way, cost of poor quality is viewed holistically and refers to all quality-related expenses, not only “poor quality” or “failures.” For instances, companies must analyze the quality checks that exist in their systems and determine if they are really essential and cost justified.
To more accurately reflect this different focus, consider using the phrase “cost of poor process and product quality,” or C3PQ for short.
The table below shows a four-bucket framework for how costs can be categorized and examples of what specifically can be measured.
| Four-bucket Framework for Categorizing and Measuring | |
| C3PQ Category | Metrics |
| Prevention |
|
| Appraisal |
|
| Internal Failure |
|
| External Failure |
|
| Soft Costs (Another Cost Measure) | Lost sales due to poor customer perception |
All of these components can represent significant cost savings opportunities that would not necessarily be uncovered by traditional LSS techniques, because the best practices of less-regulated industries that have been using LSS for a longer time cannot be directly applied to the manufacturing processes in life sciences. Those implementations should not happen until after the C3PQ costs have been both measured and controlled.
The first step in making improvements is to work with the finance department to develop a baseline for measuring current-state costs, whether through cost of goods supplied, labor plus material costs, etc. Only then can improvement goals be set and C3PQ metrics be reported on a regular basis.
Once costs are being measured within the four-bucket framework (and eventually with more granularity), it will become apparent for each life sciences company where its quality costs are the highest. Other relevant operational metrics should then be measured in terms of their impact on each quality cost bucket. This will enable the business to identify specific improvement targets, and to track how improvements relate to drops in C3PQ.
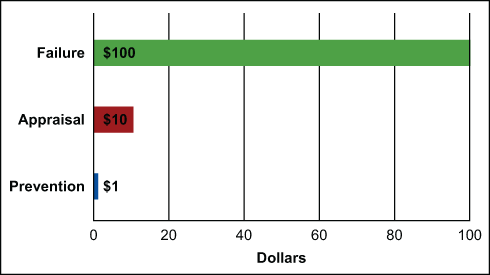
There is a need to view quality costs holistically to ensure that overall product quality is not impacted. As the focus is transferred to building-in quality rather than testing-in, however, costs will move away from failures and toward prevention. The figure below illustrates the 1-10-100 rule, which has proven true for quality costs and states that $1 properly spent in defect prevention is equal to $10 spent in product appraisal and $100 spent on product failures.
A simple shift in how existing metrics are viewed can open up a new world of cost-saving opportunities. Once the low-hanging fruit has been identified, life sciences will have no choice but to catch up with other industries in terms of using technology to ensure product quality while minimizing operational costs.
Tools such as factory automation will enable companies to continue to drive down all quality-related costs. Implementing manufacturing execution systems and advanced planning systems will deliver actionable intelligence that can be used to run the business more efficiently and consequently lower C3PQ.
Maximizing Resources
Life sciences must substantially drive down operational costs to have any hope of maintaining a level of financial success comparable to what they have experienced in the past. As other industries have adapted to changing marketplaces and improved how they conduct business, life sciences must also find an effective way to maximize how resources are utilized.
This industry would be best served to follow LSS best practices from other industries where possible, while drastically increasing the visibility of their quality-related expenditures by launching a C3PQ strategic initiative.